




Démarches administratives pour le lancement d’une évaluation aléatoire aux États-Unis
Summary
Cette check-list contient des recommandations relatives aux démarches logistiques et administratives qui sont nécessaires pour mener à bien une évaluation aléatoire respectant les dispositions légales, les règles de transparence imposées par de nombreuses revues universitaires et les procédures de sécurité imposées par les normes réglementaires ou éthiques. Nombre de ces démarches nécessitent une planification approfondie dès le début du processus de recherche. L'ordre dans lequel elles doivent être effectuées peut varier d'un projet à l’autre. Cette liste n’est donc pas nécessairement chronologique, et de nombreuses démarches sont interdépendantes.
Main relationships between the administrative steps
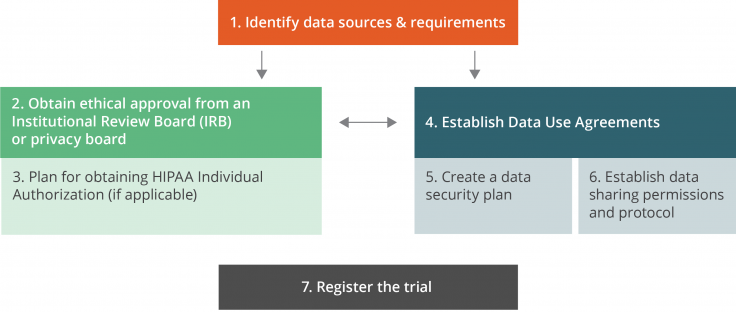
1. Identifier les sources et les besoins en matière de données
Si les données nécessaires à l’évaluation proviennent de bases administratives, cela peut contribuer à réduire les frais d’acquisition des données et limiter certains risques de biais par rapport aux données primaires. Les chercheurs doivent toutefois être conscients des implications en matière de qualité, de limites, de diversité, d'équité et d’inclusion, et bien connaître les réglementations qui régissent l’utilisation de ces données. Ils doivent également se demander si les individus qui participent à l'évaluation figurent bien dans les données. Il sera probablement nécessaire de faire une demande d'autorisation d'utilisation des données (voir l’étape 4 pour plus de détails) et de mettre au point une stratégie viable pour l’appariement des données et de l’échantillon d’étude. La procédure d’obtention d’une autorisation pour l’utilisation et l’appariement des données peut prendre du temps, et il y a parfois un décalage entre le moment où un événement se produit et celui où il est pris en compte dans les données. Les chercheurs doivent donc adapter le calendrier de lancement de leur évaluation en conséquence. Les réglementations qui s'appliquent dépendent généralement du sujet ou de la source des données administratives : les données sur l’éducation peuvent ainsi être soumises au Family Educational Rights and Privacy Act (FERPA), les données de santé peuvent être soumises au Health Insurance Portability and Accountability Act (HIPAA), et les données provenant de certains États américains relèvent par ailleurs de la législation propre à ces États.
Concernant les données primaires, leur collecte nécessite un travail de préparation qui inclut notamment la création des instruments de collecte (enquêtes, marqueurs biologiques, observation directe, applications de géographie spatiale, etc.), la validation de ces instruments par des tests sur le terrain, la formation des équipes chargées de la collecte et la mise en place d’un dispositif de supervision et de suivi continus pour garantir la fiabilité et l’homogénéité de la collecte des données.
Le niveau d’identifiabilité des données administratives, ainsi que la sensibilité et la nature des informations obtenues lors de la collecte de données primaires, ont une incidence sur les exigences en matière d’approbation éthique, d’accords d’utilisation des données, de consentement éclairé et de sécurité des données. Si les chercheurs parviennent à estimer précisément le degré de sensibilité des données dont ils ont besoin, il leur sera plus facile d’évaluer la durée et le niveau d’exigence de la procédure d’approbation.
Pour plus d’informations, consultez le Catalogue des ensembles de données administratives de J-PAL Amérique du Nord, ainsi que la ressource « Utiliser des données administrative dans le cadre d’une évaluation aléatoire », en particulier la sous-section « Biais potentiels » dans la section intitulée « Pourquoi utiliser des données administratives ? ».
2. Obtenir l’avis favorable d’un Institutional Review Board (IRB) ou d’un comité de protection de la vie privée (Privacy Board)
Toute recherche impliquant des sujets humains ou des données individuelles sur des êtres humains vivants est susceptible1 de devoir être examinée par un Institutional Review Board (IRB), même en l’absence d’interaction directe entre les sujets et les chercheurs. Suite à l’examen du dossier, le comité peut conclure que le projet de recherche 1) n'est pas une étude sur des sujets humains et n'est donc pas soumis à un examen plus approfondi, 2) est dispensé d’examen continu, 3) peut faire l’objet d’un examen accéléré par un administrateur de l’IRB ou 4) doit être examiné par le comité de l’IRB au complet. Ces décisions sont prises par l’IRB, et non par l’équipe de recherche, en fonction de l’impact potentiel de l’étude sur la santé et le bien-être des participants à l’étude.
Si plusieurs établissements dotés d’un IRB participent à l’étude (par exemple, lorsque les co-chercheurs sont affiliés à des universités différentes ou qu’un partenaire de mise en œuvre a son propre IRB), les chercheurs doivent soit obtenir l’approbation individuelle de chaque IRB, soit demander un IRB Authorization Agreement (IAA), qui permet à un établissement de transférer la responsabilité de l’examen éthique du projet, de son approbation et de son contrôle régulier à un autre établissement. Il est recommandé d’utiliser ce dispositif pour les projets de recherche impliquant plusieurs organismes dotés d’un IRB, car la procédure d’examen du dossier par l’IRB est beaucoup plus rapide lorsqu’un seul IRB est désigné comme examinateur principal.
Il est probable que l’IRB exige que tous les chercheurs et membres du personnel de l'étude qui seront amenés à interagir directement avec les participants (par exemple, pour recueillir leur consentement ou procéder à leur inscription à l’étude) ou qui auront accès à des informations d’identification personnelle suivent une formation à la recherche sur les sujets humains, comme le programme Collaborative Institutional Training Initiative (CITI).
Dans le cadre d’une demande d’approbation auprès d’un IRB, les chercheurs doivent décrire leur protocole de recherche et l’intervention de façon à permettre à l’IRB de procéder à un examen approfondi. En outre, tous les documents utilisés pour collecter des données (y compris les questionnaires ou enquêtes, ou bien les accords d’utilisation des données permettant d’acquérir des données administratives) ou pour recruter des participants (y compris les documents promotionnels) doivent être examinés et approuvés.
Une fois l’approbation obtenue, les protocoles qui n’ont pas été exemptés sont soumis à un examen continu. Cela implique notamment de notifier l’IRB de l’arrivée de tout nouveau membre du personnel, de toute modification apportée aux procédures de l’étude ou aux enquêtes et de tout événement indésirable. Si vous avez des questions concernant les éléments à signaler ou à inclure dans vos rapports annuels, nous vous invitons à vous rapprocher de l’IRB compétent.
Pour plus d’informations, consultez la section « Conformité » et l’annexe « Consentement et autorisation » de la ressource « Utiliser des données administratives dans le cadre d’une évaluation aléatoire ».
Calendrier
La plupart des IRB requièrent au moins un mois pour examiner les dossiers, et beaucoup recommandent de prévoir une période d’examen plus longue. L’université de Harvard, par exemple, suggère de soumettre les demandes au moins deux mois avant la date prévue pour le début de l'étude.
Considérations supplémentaires
Les IRB demandent souvent aux chercheurs de fournir un accord signé d’utilisation des données (DUA) avant d’approuver pleinement le protocole expérimental, tandis que les fournisseurs de données exigent souvent l’approbation de l’IRB avant de conclure un DUA. Ces deux procédures peuvent impliquer de longs délais de traitement, et les modifications apportées par une entité doivent ensuite être examinées et approuvées par l’autre. Pour résoudre ce problème, les chercheurs sont encouragés à communiquer avec chaque entité de façon proactive. Par exemple, un IRB peut être disposé à accorder au projet de recherche une approbation provisoire, l’approbation finale étant soumise à la conclusion d’un DUA. Pour plus d'informations sur les accords d'utilisation des données, voir l’Etape 4, Conclure des accords d’utilisation des données.
3. Procédure d’obtention d’une autorisation individuelle en vertu de l’HIPAA
Pour obtenir des données auprès de certains organismes de soins de santé ou liés au secteur de la santé, il est parfois nécessaire de disposer d’un document signé attestant que la personne concernée a donné son autorisation. Cela concerne notamment, mais pas uniquement, les données contenant des informations d'identification personnelle qui sont considérées comme des données de santé protégées (Protected Health Information, ou PHI) en vertu de la loi HIPAA. Cette loi impose des sanctions pénales et/ou civiles aux personnes qui utilisent ou partagent des données de façon abusive. Ces sanctions s’appliquent aux fournisseurs de données, et peuvent également s’appliquer aux chercheurs qui se sont procuré ces données. C’est pourquoi les fournisseurs de données prennent de nombreuses précautions avant de communiquer les données aux chercheurs.
Certaines exigences en matière d’autorisation peuvent être levées ou modifiées à la discrétion de l’IRB, sous réserve de justification adéquate de la part du chercheur. Dans de nombreux cas, les procédures de recueil de l’autorisation et du consentement éclairé peuvent être réalisées conjointement.
Pour plus d’informations, consultez la section « Conformité » et l’annexe « Consentement et autorisation » de la ressource « Utiliser des données administratives dans le cadre d'une évaluation aléatoire ». La procédure de recueil de l’autorisation individuelle présente de nombreux parallèles avec celle du consentement éclairé exigé par les IRB. Des conseils concernant la mise au point d’une procédure de recueil du consentement figurent dans la ressource « Définir la procédure d’admission et de recueil du consentement » du présent guide pratique.
Considérations supplémentaires
Bien que certains éléments du formulaire d’autorisation individuelle soient obligatoires, de légères modifications du texte écrit ou du style de communication de l’employé chargé d’accompagner les sujets de recherche potentiels dans le cadre de la procédure d’autorisation peuvent avoir une incidence sur le taux de participation et la conformité. Veillez à former rigoureusement les personnes chargées d’accompagner les sujets potentiels tout au long de la procédure d’autorisation et envisagez de tester au préalable différentes méthodes d’explication de cette procédure dans le cadre d’un test pilote. Des conseils en matière de formation figurent dans la sous-section « Qui va procéder au recrutement ? » de la ressource « Définir la procédure d'admission et de recueil du consentement » de ce guide pratique.
4. Conclure des accords d’utilisation des données
Pour accéder aux données d’un autre organisme, il est souvent nécessaire de conclure un accord d’utilisation des données (DUA) qui fixe les conditions de partage des données par le fournisseur et d’utilisation de ces données par le chercheur. Les DUA, qui doivent généralement être approuvés par le conseiller juridique de l’établissement d’origine du chercheur, contiennent souvent des restrictions en matière d’utilisation des données, des exigences de sécurité et des obligations de publication qui peuvent avoir un impact significatif sur la recherche concernée ou sur la liberté académique.
De nombreuses universités disposent d’un modèle type d’accord contenant des termes et conditions que l’université juge acceptables et qui tiennent compte des besoins des chercheurs. L’utilisation d’un modèle préapprouvé peut simplifier la procédure d'examen du dossier au sein de l’établissement qui a créé le modèle.
Pour plus d’informations, notamment sur les éléments essentiels à prendre en compte lors de l’examen d’un DUA et sur la négociation de ces accords, voir les sections « Si nécessaire, établir un contrat de recherche » et « Accords d’utilisation des données » dans la partie « Formaliser le partenariat de recherche et définir les rôles et les attentes » de ces ressources.
Calendrier
Dans une analyse de 2015 sur les démarches d’acquisition de données auprès de 42 agences de données, MDRC a constaté qu’il s'écoule généralement 7 à 18 mois entre la première prise de contact avec un fournisseur de données et la conclusion d’un accord juridique.2 Une grande partie de ce temps est consacrée à la double procédure permettant d’obtenir l’approbation de l’IRB et l’autorisation légale pour la demande de données. Les IRB demandent souvent aux chercheurs de fournir un DUA signé avant d’approuver le protocole expérimental, tandis que les fournisseurs de données exigent l’approbation de l’IRB avant de signer un DUA. Les deux procédures peuvent impliquer de longs délais de traitement, et les modifications apportées par une entité doivent être examinées et approuvées par l’autre.
5. Élaborer un plan de sécurité des données
Tout ensemble de données contenant des informations d’identification personnelle doit être protégé, en particulier s’il contient également des informations potentiellement sensibles. Les IRB, les fournisseurs de données (par exemple les Centers for Medicare & Medicaid Services) et certains organismes de financement (par exemple les National Institutes of Health) demandent aux chercheurs de fournir une description de leurs procédures de sécurité des données. Il s’agit notamment de décrire les modalités de contrôle de l’accès des utilisateurs, les politiques en matière de partage des données, de chiffrement ou de protection par mot de passe, ainsi que les possibilités de réidentification des données désidentifiées.
Un aspect important de la sécurité des données consiste à veiller à ce que les données soient correctement stockées. Mettez en place un plan de prévention de la perte des données essentielles, y compris les données brutes, les listes d’assignation de traitement et les tableaux de correspondance entre les identifiants pseudonymisés de l'étude et les informations d’identification personnelle. Ces données doivent être régulièrement sauvegardées dans au moins deux endroits distincts et sécurisés.
Certains projets nécessitent la création de nouveaux emplacements de stockage des données, tels qu’un serveur hébergé par l’établissement, un ordinateur hors ligne conservé dans un lieu sécurisé ou, plus simplement, un dossier sur un site de stockage cloud (comme Box, Dropbox ou Google Drive). Le choix de l’emplacement de stockage dépend des exigences en matière de sécurité des données et des dispositions de l’accord d’utilisation des données.
Pour plus d’informations, consultez notre ressource « Procédures de sécurité des données pour les chercheurs »
6. Obtenir les autorisations de partage des données et mettre au point le protocole de partage
J-PAL et un certain nombre d’organismes de financement, dont la National Science Foundation et les National Institutes of Health, ont adopté des politiques de partage des données pour les recherches qu’ils financent. De nombreuses revues scientifiques de premier plan imposent en outre le partage des données et du code de réplication comme condition préalable à la publication (c’est notamment le cas de la American Economic Review, d’Econometrica, et de Science.)
Créer et stocker des ensembles de données et des fichiers de code en vue d’une publication future évitera aux chercheurs de perdre trop de temps à préparer les données à un stade ultérieur du processus de recherche.
Pour publier des données, même désidentifiées, il peut être nécessaire d’obtenir l’autorisation des fournisseurs de données, qu’il s’agisse d’individus, d’agences ou d’entreprises. Il est possible que l’accord d’utilisation des données doive inclure des dispositions autorisant la publication des données, ce qui peut influer sur le niveau de désagrégation autorisé. Les comités d’examen peuvent en outre exiger que les procédures de recueil du consentement éclairé et/ou de l’autorisation HIPAA mentionnent également tout projet de publication des données.
Même lorsque les données ne peuvent être partagées du fait de leur caractère sensible ou en vertu d’un accord d’utilisation des données, il est quand même possible de partager le code de réplication, les métadonnées, les livres de codes et les procédures d’accès aux données et d’appariement pour permettre à d’autres chercheurs de comprendre et éventuellement de répliquer votre travail. Faire en sorte de partager les données autant que possible afin de favoriser la transparence et la reproductibilité de la recherche en sciences sociales est une excellente pratique.
Pour plus d’informations, consultez les ressources de J-PAL sur la transparence et la reproductibilité.
7. Enregistrer l’essai
L’enregistrement sur clinicaltrials.gov est requis par la FDA pour de nombreux essais médicaux, cliniques ou en rapport avec la santé. Les NIH exigent également que tous les essais cliniques qu’ils financent soient enregistrés sur clinicaltrials.gov et que leurs résultats y soient publiés. Les NIH utilisent une définition large de l’essai clinique, qui inclut les évaluations d’interventions non réglementées par la FDA, y compris les interventions comportementales étudiées en sciences sociales. Certaines revues médicales exigent l’enregistrement sur clinicaltrials.gov comme condition préalable à la publication d’un article, et ce même si la réglementation de la FDA ou des NIH ne s’applique pas.
L’enregistrement sur d’autres sites, tels que le RCT Registry de l’American Economics Association (soutenu par J-PAL) ou l’OSF, peut être exigé par les bailleurs de fonds des projets, y compris J-PAL. L’enregistrement des essais permet de garantir la transparence de la recherche. Les registres d’essais cliniques constituent une source de résultats scientifiques qui peuvent servir à réaliser des méta-analyses, mais aussi à repérer les instruments d’enquête et les données disponibles.
L’enregistrement de l’essai doit être effectué avant le lancement de l’intervention étudiée.
Dernière modification : septembre 2021.
Ces ressources sont le fruit d’un travail collaboratif. Si vous constatez un dysfonctionnement, ou si vous souhaitez suggérer l'ajout de nouveaux contenus, veuillez remplir ce formulaire.
Ce document a été traduit de l’anglais par Marion Beaujard.
Nous remercions Amy Finkelstein et Pauline Shoemaker pour leurs idées et leurs conseils précieux. Ce document a été traduit de l’anglais par Marion Beaujard. Le présent travail a pu être réalisé grâce au soutien de la Fondation Alfred P. Sloan et d'Arnold Ventures.
Additional Resources
Catalogue des ensembles de données administratives| https://www.povertyactionlab.org/admindatacatalog
Mesures et collecte des données| https://www.povertyactionlab.org/research-resources/measurement-and-data-collection
Transparence et reproductibilité| https://www.povertyactionlab.org/research-resources/transparency-and-reproducibility
Le catalogue des ensembles de données administratives de J-PAL Amérique du Nord répertorie un certain nombre d’ensembles de données clés aux États-Unis et en décrit les procédures d’accès sur la base des informations fournies par les agences concernées.
La ressource « Utiliser des données administratives dans le cadre d'une évaluation aléatoire », élaborée par J-PAL Amérique du Nord, offre des conseils généraux et pratiques pour identifier les sources de données administratives, en évaluer la qualité et le contenu, connaître les exigences en vigueur et obtenir et exploiter des données administratives non publiques dans le cadre d’une évaluation aléatoire.
Le document “The Lessons of Administrative Data” présente des exemples d’études de premier plan qui ont pu être réalisées grâce à des données administratives.
Des ressources sur la collecte de données primaires sont disponibles sur le site de J-PAL.
Des informations sur les systèmes de données intégrés sont disponibles sur le site Actionable Intelligence for Social Policy.
Le ministère américain de la santé et des services sociaux, les Centers for Disease Control and Prevention et le National Center for Health Statistics proposent une boîte à outils sur les pratiques responsables en matière de collecte, d’utilisation, de protection et de partage des données relatives à la santé.
Des chercheurs de l'université de Harvard travaillent à l’élaboration d’outils pour aider les chercheurs à déterminer quelles réglementations s’appliquent aux données qui les intéressent par l’intermédiaire d’un questionnaire interactif. Les Institutional Review Boards, les bibliothécaires de données et les services responsables de la conformité peuvent également fournir une aide précieuse pour déterminer quelles réglementations s’appliquent.
Les IRB examinent les protocoles expérimentaux en se basant sur les exigences de la Common Rule (45 CFR 46.116). Se référer à ces règles et rédiger les protocoles et les dossiers de demande d’avis en les gardant à l’esprit peut favoriser la communication entre l'équipe de recherche et l’IRB et augmenter les chances de réussite du dossier.
Le Committee on the Use of Humans as Experimental Subjects (COUHES) du Massachusetts Institute of Technology (MIT) donne des exemples de différents types de formulaires de demande d’avis et fournit également des instructions à ce sujet.
Le COUHES propose également des lignes directrices pour plusieurs étapes de la procédure d’examen du projet par l'IRB.
Le Committee on the Use of Human Subjects (CUHS) de l’université de Harvard propose d’autres exemples de formulaires de demande d’avis.
L’université du Colorado a rédigé plusieurs guides abordant les différents aspects de la procédure des IRB.
Le ministère américain de la santé et des services sociaux propose des diagrammes d’aide à la prise de décision pour comprendre les exigences de l’IRB.
Les National Institutes of Health mettent à disposition des exemples de protocoles, dont un conçu pour les chercheurs en sciences sociales et comportementales, qui sont disponibles dans l’outil de rédaction de protocoles en ligne des NIH.
45 CFR 164.502 – Uses and disclosures of protected health information (texte officiel).
45 CFR 164.508 – Uses and disclosures for which an authorization is required (texte officiel). Réglementation HIPAA relative aux autorisations de divulgation d’informations de santé et aux conditions requises pour une telle autorisation.
Lignes directrices des NIH sur le respect de la loi HIPAA, portant notamment sur les autorisations de recherche et les dispenses d’autorisation.
Le guide du ministère américain de la santé et des services sociaux sur le lien entre la règle de confidentialité de l’HIPAA et la recherche décrit notamment les conditions spécifiques à remplir pour obtenir une autorisation de recherche.
De nombreuses universités disposent d’un modèle type d’accord contenant des termes et conditions que l’université juge acceptables et qui tiennent compte des besoins des chercheurs. L’utilisation d’un modèle préapprouvé peut simplifier la procédure d’examen du dossier au sein de l’établissement qui a créé le modèle.
- Le MIT met à disposition des exemples d’accords d’utilisation des données et d’accords de confidentialité ici.
De nombreuses universités de recherche proposent une assistance et des conseils en matière de sécurité des données par l’intermédiaire de leur service informatique et du personnel spécialement chargé de l’informatique au sein de leurs départements académiques. Les chercheurs qui mettent en place des mesures de sécurité des données sont invités à consulter le personnel du service informatique de leur établissement d’origine, qui pourra leur recommander des logiciels de sécurité spécifiques et leur apporter un soutien technique.
Les fournisseurs de services de stockage cloud offrent toute une gamme d’options de sauvegarde des données et proposent parfois des solutions supplémentaires pour sauvegarder les données pendant des périodes plus longues afin de se prémunir contre tout effacement accidentel. Les serveurs des établissements disposent parfois de dispositifs de sauvegarde des données, et il existe également des systèmes de sauvegarde au niveau des appareils. La sauvegarde des données sur un disque dur externe (stocké dans un lieu distinct des ordinateurs utilisés au quotidien) constitue une autre solution pour les environnements à faible connectivité.
Pour les chercheurs au MIT, le Dr Micah Altman, directeur de la recherche des bibliothèques du MIT, donne régulièrement des conférences sur la gestion des données confidentielles.
Le Département des systèmes d’information et des technologies du MIT propose des ressources sur les thèmes suivants (identifiants MIT requis) :
- Protéger les données
- Secure Shell File Transfer Protocol: SecureFX
- Chiffrement (y compris des recommandations de logiciels) et chiffrement complet du disque
- Supprimer les données sensibles
La Research Data Security Policy (HRDSP) de l’université de Harvard est une excellente ressource pour la classification des différents niveaux de sécurité. Elle donne également des exemples d’exigences en matière de sécurité.
J-PAL propose des ressources et des liens vers des sources d’information supplémentaires sur la transparence et la reproductibilité.
La Berkeley Initiative for Transparency in the Social Sciences (BITSS) est une excellente source d’informations en matière de reproductibilité et de publication des données. Elle organise fréquemment des sessions de formation en présentiel. Son Manual of Best Practices in Transparency in Social Science Research offre des suggestions pour rédiger un code d’analyse reproductible dès le départ.
Innovations for Poverty Action (IPA) met à disposition des ressources sur la transparence de la recherche. Les chercheurs affiliés à IPA peuvent en outre bénéficier d’un soutien supplémentaire pour la conservation de leurs données et la vérification de leur code. Le manuel intitulé Best Practices for Data and Code Management aborde les principes relatifs à l’organisation et à la documentation des données à toutes les étapes du cycle de vie du projet dans le but de rendre la recherche reproductible.